
— Étude collaborative du CDC du Zhejiang, de Macro & Micro-Test et du CDC chinois publiée dans Frontiers in Cellular and Infection Microbiology
Aperçu de l'étude
En mai 2026, la revue Frontiers in Cellular and Infection Microbiology (JCR Q1, IF ≈ 4,6) a publié un article dirigé par le Centre provincial de contrôle et de prévention des maladies du Zhejiang (CDC du Zhejiang), en collaboration avec l'équipe de bioinformatique de Beijing Macro & Micro-Test Bio-Tech Co., Ltd. et l'Institut national de contrôle et de prévention des maladies transmissibles (CDC de Chine). L'étude est intitulée :
« Identification et analyse phylogénétique de sept souches de Brucella abortus dans le Zhejiang, en Chine. »

Cette étude représente la première analyse phylogénétique systématique et complète du génome de Brucella abortus (B. abortus) dans la province du Zhejiang, en Chine. L’équipe a analysé sept isolats collectés entre 2015 et 2025 (quatre souches d’origine humaine et trois d’origine bovine provenant de Jinhua, Quzhou et Ningbo). Les résultats apportent des preuves génomiques quant à l’origine et aux voies de transmission de cette espèce dominante du nord dans une région épidémique atypique du sud de la Chine orientale.
Contexte et importance
La brucellose est une zoonose causée par des bactéries du genre Brucella. Brucella abortus infecte principalement les bovins, mais peut également provoquer la maladie chez l'homme. En Chine, la brucellose présente une forte variation géographique : l'incidence la plus élevée se situe dans les provinces du nord (par exemple, la Mongolie-Intérieure, le Shanxi et le Heilongjiang). À l'inverse, les provinces du sud, notamment le Zhejiang, ont historiquement été dominées par Brucella melitensis, avec très peu de cas de B. abortus recensés. Cette disparité régionale fait de la caractérisation génétique et de la recherche de la source de B. abortus au Zhejiang une priorité majeure de santé publique.
Méthodes et principaux résultats
L'équipe de recherche a adopté une stratégie à plusieurs volets combinant biologie moléculaire et bioinformatique :
1.Identification et typage de base des agents pathogènes
Les tests PCR du gène BCSP-31 et AMOS-PCR ont confirmé que les sept isolats étaient tous des B. abortus.
Le typage de séquence multilocus (MLST) basé sur neuf gènes de ménage a révélé que tous les isolats appartenaient au type de séquence ST2, indiquant une forte homogénéité génétique parmi les souches de B. abortus circulant dans le Zhejiang.
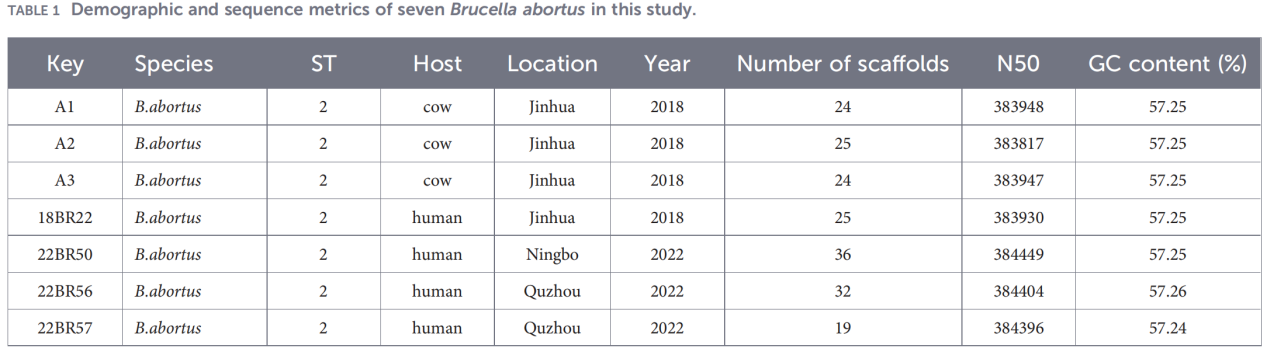
2.Caractérisation du génome entier
Le séquençage du génome entier a été réalisé sur la plateforme Illumina NovaSeq. L'analyse de l'identité nucléotidique moyenne (ANI) a montré que les isolats du Zhejiang présentaient une similarité allant jusqu'à 99,99 % avec la souche de référence B. abortus 544.
L'analyse du pan-génome a révélé une population hautement conservée : 3 084 gènes de base ont été identifiés, ainsi que seulement 10 gènes de coquille, et aucun gène de noyau mou ou de nuage n'a été détecté.
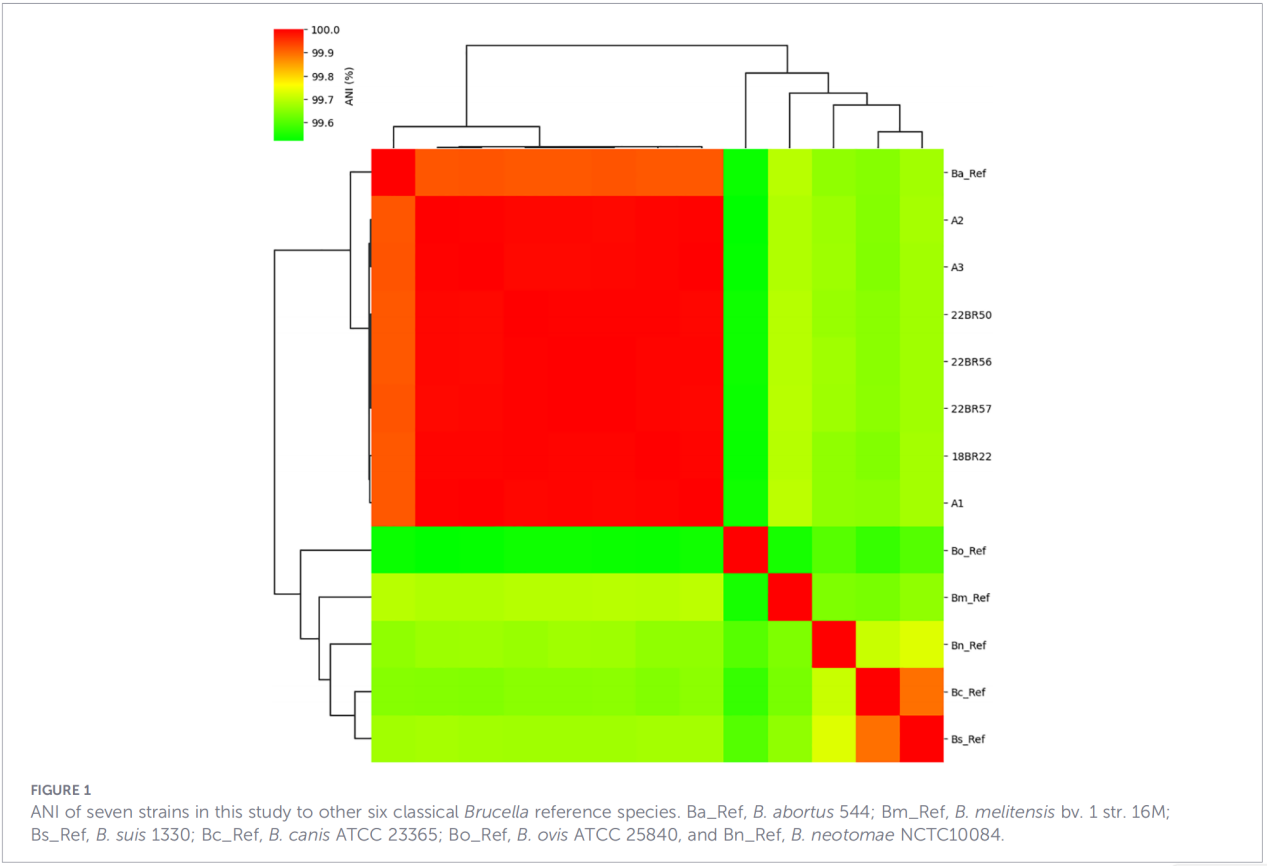
3.Profils génétiques de virulence et de résistance aux antimicrobiens
Au total, 68 facteurs de virulence ont été prédits, couvrant des voies classiques telles que la biosynthèse des LPS, le système de sécrétion T4SS et le système de régulation à deux composants BvrR-BvrS. Notamment, tous les isolats étaient dépourvus des gènes d'adhésine bmaA et btaF. L'analyse des gènes de résistance n'a détecté que le gène mprF dans la base de données CARD, aucun autre déterminant de résistance n'ayant été identifié.
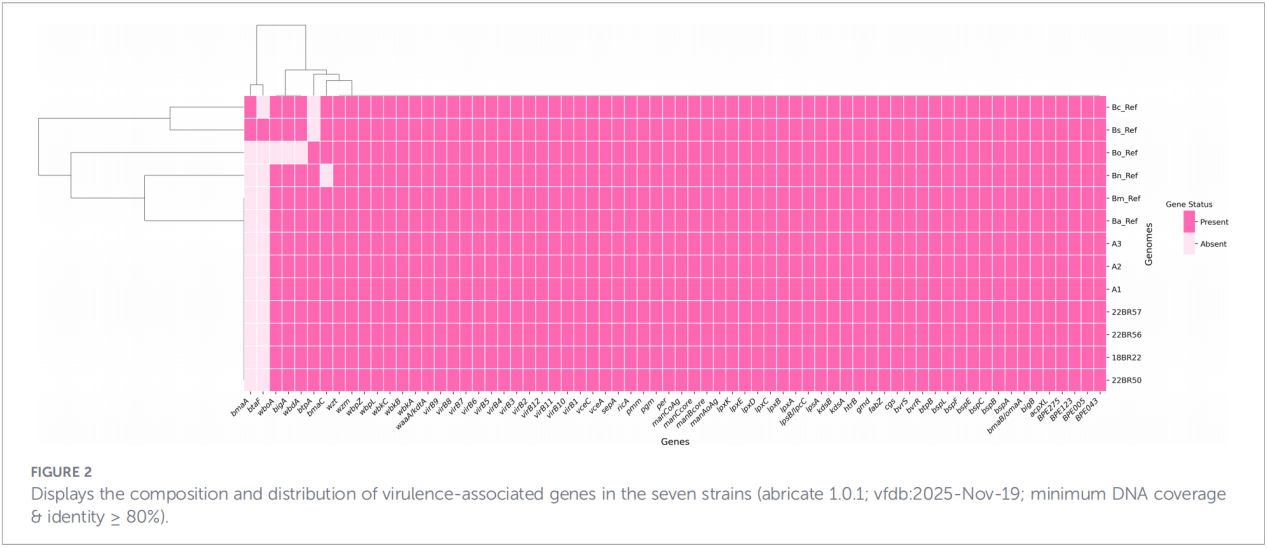 4. Reconstruction phylogénétique et traçage de la transmission
4. Reconstruction phylogénétique et traçage de la transmission
L'analyse des polymorphismes nucléotidiques simples du génome central (cgSNP) a permis de positionner les isolats du Zhejiang à un endroit précis de l'arbre phylogénétique mondial. Les résultats ont montré que les souches du Zhejiang forment un groupe monophylétique avec des souches provenant de Russie, de Mongolie et de plusieurs provinces du nord de la Chine (Ningxia, Heilongjiang, Mongolie-Intérieure, Hebei, Gansu, Pékin). Ce groupe se subdivise en trois sous-clades distincts (clades 1 à 3), suggérant de multiples introductions indépendantes.
Conclusions et implications
Cette étude fournit le premier ensemble de données génomiques de haute précision de B. abortus dans la province du Zhejiang et aboutit à plusieurs conclusions clés :
- Cléar fond génétique– Les souches de B. abortus circulant dans le Zhejiang appartiennent au ST2, sont génomiquement hautement conservées et représentent une lignée typique de brucellose bovine.
2. Evidengue de transmission interrégionaleL’analyse phylogénétique ne confirme pas l’existence d’une lignée endémique indépendante dans le Zhejiang. Au contraire, les données suggèrent fortement que ces souches proviennent du nord de la Chine et pourraient partager une origine évolutive commune avec des souches de Russie et de Mongolie. La présence de trois sous-clades implique plusieurs introductions distinctes.
3. implications pour la santé publiqueCes résultats soulignent l’importance de la surveillance génomique de la brucellose, même dans des régions traditionnellement non endémiques comme le Zhejiang. Bien que le nombre de cas soit actuellement faible, des outils à haute résolution tels que le cgSNP permettent de retracer efficacement la source des foyers importés et de fournir des preuves scientifiques pour interrompre les chaînes de transmission liées au transport interprovincial de bétail.
Ce travail comble non seulement une lacune en matière de recherche dans la province du Zhejiang, mais fournit également de nouvelles données de référence pour la surveillance des agents pathogènes et l'évaluation des risques de brucellose dans la région du delta du Yangtsé.
Informations sur le document :
Yang, Y., Shi, X., Chen, J., Wang, L., Wu, Z., Yao, W., … & Wu, B. (2026). Identification et analyse phylogénétique de sept souches de Brucella abortus dans le Zhejiang, en Chine. Frontiers in Cellular and Infection Microbiology, 16, 1758965.
Date de publication : 10 juin 2026